
What is Corticobasal Degeneration
Corticobasal degeneration, otherwise termed as, Corticobasal ganglionic degeneration or CBGD, is a kind of neurodegenerative disorder concerning a certain protein, called tau. In the disorder, there is an abnormal or unhealthy assembly of the tau protein in an individual’s brain.
This phenomenon is medically referred to as tauopathy that ultimately leads to the deterioration or the degeneration of the individual’s brain. Generally, the main parts of the brain that are directly involved are the basal ganglia and the cerebral cortex. Moreover, corticobasal degeneration is also a progressive disorder; meaning, it worsens as time passes by.
As an added information, a group of physicians led by doctor Rebeiz in the late 1960s first observed the occurrence of corticobasal degeneration in three affected people exhibiting the hallmark characteristics of the disorder.
Epidemiology
Corticobasal degeneration is a rather uncommon disease and usually occurs in people in their advanced age, usually in the sixth decade of life. However, the youngest age recorded to have had the disease was at 28 years old. Both sexes, males and females may be affected by the disorder; however, the disorder is more likely to occur in the female population more than the male population.
Recent studies show that the disorder’s prevalence most likely fall under a 5 to 7 per 100,000 individual rate and have multiplied in great amounts since the disorder’s discovery. This multiplication in number is indicative of a more sensitive diagnosis or simply, corticobasal degeneration is more frequently occuring in the present days.
Moreover, corticobasal degeneration has no known racial partiality; meaning, it can occur equally through different races.
Causes
The definite cause of corticobasal degeneration has not yet been fully elucidated; however, some reports have identified familial or genetic factors. The particular gene of interest that has been the focus of research studies, associated with corticobasal degeneration is the gene in chromosome 17.
Pathology
Corticobasal degeneration affects both the subcortical as well as the cortical parts of the human brain. Moreover, glial cells as well as nonpyramidal neurons and pyramidal neurons are also affected. As mentioned above, corticobasal degeneration is characterized by an abnormality called tauopathy. Presently, the disorder is categorized under the classification 4-repeat tauopathy.
This distinction is the main factor that sets corticobasal degeneration apart from its similar diseases such as Alzheimer’s disease and progressive supranuclear palsy. The basal ganglia and the cortex of the brain are characterized by puffed-up neurons as well as achromasia or the lack of staining in the cytoplasmic area. In addition, a loss of neurons both in the cortical as well as in the subcortical area may also occur due to the disease. In addition, histologic findings reveal atrophy in the frontoparietal area as well as entanglement of neurofibrils.
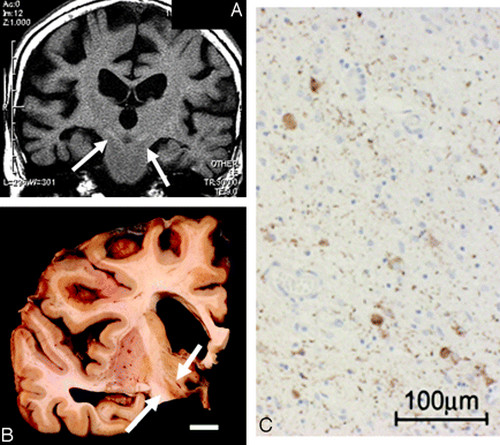
Picture : Corticobasal degeneration in 84 year old woman
Note : A: Coronal T1-weighted image B: Macroscopic specimen (subthalamic nuclei showing brownish changes) C: Microscopic examination (gliosis is seen)
Symptoms and Signs
Corticobasal degeneration may be categorized into two. It may be a syndrome of motion problems as well as cognitive difficulty, or it may be a disease entity per se. Regardless, these motion and cognitive problems are usually utilized as early indicators that the disease is present. Moreover, motion problems are usually assymetrical in nature; meaning, what happens in one side of the body won’t necessarily happen on its counterpart. For example, a motion problem on the right hand may not necessarily occur in the left hand and vice versa.
In addition there are four signs and symptoms (both motion and cognitive) that are major characteristics associated with corticobasal degeneration. These signs and symptoms are apraxia, alien hand syndrome, aphasia and parkinsonism. Each of these four characteristics will be further discussed below.
Apraxia
Apraxia is a condition wherein an individual loses the ability to perform tasks that are normally familiar and are previously done without difficulty. In the case of corticobasal degeneration, ideomotor apraxia most likely occurs; this apraxia may affect both of the upper and lower extremities. For example, an individual may have difficulty to initiate a simple act of walking, as both feet may seem to be glued in the ground. In addition, apraxia may also occur and affect fine motor movements normally performed by the hands as well as the digits.
Alien hand Syndrome
Alien hand syndrome is fairly common in people with corticobasal degeneration. From the word itself, an individual may sense a feeling of alienation from his or her own hands. This happens because the individual affected may have a difficulty, or entirely lose the ability to control his or her own hands, hence the feeling of alienation. Moreover, alien hand syndrome in corticobasal degeneration is asymmetrical in nature. As explained above, asymmetrical means that an alien hand syndrome in the right hand do not necessarily entail an alien hand syndrome in the left hand or vice versa. Lastly, alien hand syndrome do not occur without reason. It actually is caused by an outside stimulation or otherwise known as external stimuli.
Aphasia
Aphasia in corticobasal degeneration is characterized by the inability to initiate a verbal act. In other words, the individual loses the capability to speak a word or to converse in a dialogue, resulting to defected patterns of speech and essential words in a sentence necessary to form a coherent thought are being omitted. As the disease progress into its late course, the individual ultimately loses the capability to utter a word.
Parkinsonism
The most common sign and symptom of Parkinsonism in corticobasal degeneration is the rigidity of the limb. Other common signs and symptoms are various disorders of the individual’s gait and bradykinesia. As in other manifestations of corticobasal degeneration, Parkinsonism is also assymetrical in nature and most likely affects the individual’s limbs.
Other motor and cognitive signs and symptoms found in corticobasal degeneration are as follows:
- Presence of depression,
- Amnesia,
- Impairment in judgment,
- Impaired movements of the eye (usually horizontal and upgrade impairments),
- Problems in gait,
- Involuntary or uncontrolled jerking of the muscles or medically referred to as myoclonus,
- Sensory loss of a cortical origin, and;
- Spatial neglect
The presence of major hallucinations however, is indicative that the patient do not actually have corticobasal degeneration, instead the patient may have Lewy body disease (diffused).
Diagnostic Tests and Laboratory Studies
The diagnosis of corticobasal degeneration is rather uneasy to do, due to the fact that its signs and symptoms very much resembles that of other diseases such as Alzheimer’s disease, Parkinson’s disease, as well as Pick complex disease. Moreover, in order to do a correct and accurate diagnosis, experts have developed a standard set of signs and symptoms or characteristics that are present in each stage of the disease as it progresses. This is done because the disorder is progressive in nature and different signs and symptoms may occur at each stage.
As it is said, corticobasal degeneration very much resembles other diseases such as Alzheimer’s disease, Parkinson’s disease, as well as Pick complex disease. That is why it is important and essential to do differential diagnoses to separate corticobasal degeneration from the three mentioned diseases. This is done to ensure proper and appropriate medical management for the respective disorders. However, other than those mentioned diseases, differential diagnoses are also done to differentiate corticobasal degeneration from the following disorders:
- Cardioembolic stroke,
- Thalamic stroke,
- Epilepsia partialis continua,
- Huntington disease,
- Creutzfeldt-Jakob disease,
- Hydrocephalus,
- Neuroacanthocytosis,
- Wilson disease,
- Whipple disease,
- Thyroid disease, and;
- Striatonigral degeneration
Laboratory studies are also performed for various purposes such as determination of cognitive causes. These laboratory studies are the following:
- Ceruloplasmin,
- Level of B-12 in the body,
- Tests for thyroid function,
- Complete blood count, and;
- Electrolyte levels
Magnetic resonance imaging or MRI of the brain is also performed to determine the size and form of the midbrain (if corticobasal degeneration is present, the midbrain size should be fairly normal), determine cortical atrophy and evaluation of the basal ganglia, whether if it exhibits atypical signal.
Other tests performed are tests for the evaluation of the patient’s neuropsychological state as well as electroencephalography.
On the other hand, other procedures that are performed are lumbar puncture for the evaluation of the cerebrospinal fluid, jejunal biopsy, PCR test or polymerase chain reaction test to determine presence of Tropheryma whippleii and brain biopsy.
Medical Management and Treatment
Presently, there is unfortunately no definitive cure for the disorder, as its definitive cause has not yet been fully elucidated. However, essentially, the medical management for corticobasal degeneration is divided into three evaluations, which will be discussed below.
First evaluation
During the initial evaluation, the medical care focuses on alleviating or treating presenting signs and symptoms or the symptomatic approach to treatment. Discontinuation of various medications may be necessary such as ceasing anticholinergic drugs to prevent damage in memory and attention function of the individual. However, if the patient already has memory loss, patient may be prescribed with Vitamin E as well as antioxidants. Aside from that, medications that may possibly cause Parkinsonism are also ceased. Moreover, if symptoms of rigidity are already debilitating to the patient, a regimen for levodopa may be started. Also, if the dystonia of the limb are already painful, injections of botulinum toxin may be instituted.
Second evaluation
During the follow-up evaluation, or the second part of the medical management, treatment of associated systemic situations identified in the serologic examinations may be started. If in the second evaluation, rigidity is still present, indicating that the levodopa treatment is ineffective, then discontinue the regimen and institute other line of medications such as dopaminergic drugs in the third line.
Third evaluation
In the third and last evaluation, systemic situations that have presented further through the course of the disease are being recognized and treated appropriately. Modifications in dopaminergic regimen may also be done if prior dopaminergic drugs are rendered ineffective.
Moreover, the activity of the patient is not limited or restricted; however, physical assistance may be needed as the disease progresses to its late stage.
Supportive therapies
Along the course of the three evaluations, supportive therapies are done to aid in the medical management of corticobasal degeneration. Examples of these supportive therapies are as follows:
- Electroencephalography or EEG for the identification of myoclonus,
- Consultation to various specialized therapists such as speech, physical as well occupational therapists for the planning of appropriate physical activities, safety assessment and evaluation of possible assistive devices if needed for the patient,
- Management of associated dementia by providing reading materials (online materials such as the CGBD caregivers report are very helpful to families with members having corticobasal degeneration) and referring the patient and significant others to a case manager or other resource persons for the management of associated dementia,
- Spinal tap may also be done to rule out Whipple disease; however, proper evaluation and great care should be practiced since a spinal tap may cause subdural hematoma if the patient has cerebral atrophy,
- Lastly, brain biopsy may also be done if the patient and his/her significant others so desire
Diet
Patient with corticobasal degeneration may develop dysphagia or the difficulty to swallow, hence proper adjustment regarding the patient’s diet may be necessary. Depending on the extent of the dysphagia, the patient may be subjected to a soft food diet to a thickened liquids diet.
The patient may also experience constipation, hence proper diet adjustment considering constipation may also be necessary such as increase in fluid intake and intake of high-fiber foods. Medications such as laxatives and stool softeners may also be prescribed as well as promotion of the patient’s physical activity.
Follow-up
Regular follow-up should be done to monitor the patient as well as the condition’s progress and to adjust medication regimens such as dopaminergic therapies.
Prognosis
As mentioned above, corticobasal degeneration is a progressive disease, hence, both the cognitive as well as the motor problems of the patient worsens as time passes by. According to recent records, the individual affected by corticobasal degeneration has roughly eight to ten years of life from the onset of the disease.
Normally, the patient does not die due to the disorder per se, but due to the complications that comes along with the disease such as complications in the respiratory system (aspiration pnuemonia) and various infections.
References
http://en.wikipedia.org/wiki/Corticobasal_degeneration
http://emedicine.medscape.com/article/1150039-overview
